relevant product failures mainly because neither medical professionals nor people have all of the information required to make adequate judgments of item high-quality and authorized tort solutions are slow, inefficient, and intensely expensive. The modifications into the CGMP regulation would require manufacturers to extend [Webpage 52644] their good quality devices to include various new locations, for example style and getting, and to explain or extend selected present needs. Numerous of the improvements to your regulation allow it to be far more in step with ISO 9001:1994 excellent requirements. The rule will have an effect on all health care device establishments engaged in the look, manufacture, agreement sterilization, and packaging of health-related products. This Assessment presents The prices and advantages of the ultimate CGMP rule and displays the differences in between the proposed and closing regulation. The entire methodology and preliminary economic Investigation was presented while in the November 1993 ERG report, ``Economic Investigation of Proposed Revisions to The nice Manufacturing Methods Regulation for Clinical Equipment''. While the proposed rule protected ingredient manufacturers, the expense of compliance for these kinds of manufacturers was inadvertently omitted from your November 1993 ERG report. Nevertheless, FDA has made the decision not to include component manufacturers, consequently a lot of the preliminary Investigation stays valid (e.g., estimates of labor and resource specifications, level of compliance, and quantity of companies remain the exact same for the final Investigation, except wherever noted).
As well as complex facets of optimization, you will discover features associated with shopper prerequisites, such as usage of ground breaking but proven systems to supply most benefit.
FDA thinks that it's important for the individual accountable for launch to get Individually documented and dated that launch. This may be achieved by means of use of an inspection stamp, Should the stamp is managed as discussed higher than beneath Sec. 820.40 Doc controls. Hence, FDA has retained the need to get a signature. 151. Several remarks on proposed Sec. 820.eighty(e), ``Inspection and test records,'' stated that manufacturers really should not be necessary to file the usage of normal gear in inspection and exam data, due to the fact this requirement might be burdensome to massive manufacturers who use quite a few typical pieces of equipment. Some feedback stated which the file needs below Sec. 820.eighty(e) are extremely prescriptive and go very well further than ISO 9001's comparable needs. The responses mentioned that recordkeeping needs to be specified because of the manufacturer while in the spirit of ISO 9001, and should consist of just the minimal documents required to present that finished system inspections are executed in accordance with established treatments. FDA agrees that it will not be important to document every piece of apparatus Employed in acceptance routines. The prerequisite, renamed ``Acceptance information,'' now supplies that tools made use of shall be documented ``where correct.'' For many crucial functions and screening, identification from the equipment utilised might be imperative for suitable investigations into nonconforming merchandise. The necessities, as revised, are comparable to those in ISO 9001:1994. As talked about earlier mentioned, certain information and facts should be captured on acceptance records for that data to generally be beneficial in analyzing nonconformance. As a result of a few years of working experience, FDA has identified what it thinks to become a minimal requirement for these data. Part 820.eighty(e) reflects that willpower.
As a result of its risk-averse nature, the pharmaceutical marketplace has actually been slow to adopt continual processing technological innovation. It can be thrilling to find out that almost all huge pharmaceutical providers are on the forefront of early adoption. GlaxoSmithKline and Eli Lilly have declared programs to develop ongoing manufacturing crops in Singapore and Eire, respectively. Other massive companies like Novartis, Merck, Bayer, and AstraZeneca are actually focusing on steady manufacturing for a few years. A little quantity of agreement manufacturing corporations (CMO) have also specialised in constant manufacturing. The business is likely to witness a rising development in continual manufacturing of APIs, in addition to tableted products.
ii. Unit Grasp Document (DMR) (Sec. 820.181) 183. Some remarks on proposed Sec. 820.181 Gadget master report said that the prerequisite for your ``skilled'' person to get ready the DMR must be deleted since it is unclear or redundant with the requirements in Sec. 820.twenty five. FDA hasn't deleted the requirement with the DMR for being prepared, dated, and approved by a qualified particular person as the company thinks this is important to guarantee consistency and continuity in the DMR. The part is according to the initial CGMP, Sec. 820.181. FDA has, however, substituted the phrase ``geared up and authorized in accordance with Sec. 820.forty'' to be according to the requirements currently in Sec. 820.40 and also to do away with any redundancy. 184. Two opinions on Sec. 820.181(a) mentioned that ``software package layout technical specs'' should not be included in the DMR since these documents is going to be located in the DHF. One more remark requested the necessity which the DMR comprise ``software program resource code'' facts be amended due to the fact [Page 52638] resource codes for commercialized program will not be accessible to the machine manufacturers. A further comment said which the resource code shouldn't be in the DMR mainly because it will presently be during the DHF. FDA deleted the reference to ``program supply code'' due to the fact This really is by now included Along with the prerequisite for ``software specs.'' The final software specifications needs to be transferred into creation. Thus, the final software program specification for The actual machine or variety of device need to be located or referenced while in the DMR, though any earlier Variation really should be located or referenced in the DHF. FDA website believes that it's far more vital for manufacturers to build a document composition which is workable and traceable, than to worry about no matter whether anything is contained in a single file compared to An additional. The DMR is ready approximately incorporate or reference the processes and specifications which have been latest over the manufacturing floor. The DHF is meant to become more of a historic file for utilization during investigations and ongoing structure efforts.
These price savings, nonetheless, couldn't be quantified. However another good thing about the revised regulation relates to the harmonization of the final CGMP regulation While using the ISO 9001:1994 Worldwide typical. This alteration would In particular profit export-
Our optimization strategy comprises both classical resolve of proven suitable ranges (PAR) values and, in restricted collaboration with customers, methods of multivariate Examination together with other components of procedure analytical technologies.
The next stage in pharmaceutical manufacturing includes the final formulation on the medicine. The final formulation belongs towards the manufacturing sector.
The phrase “virtual audit” relates to inspections done off-internet site using enhanced conversation and information technologies to fulfill a authorized requirement of the on-web site inspection. The only real big difference would be that the inspector just isn't bodily current. These audits could also be referred to as “remote” or as “distant inspections.”
His action has always been devoted to advancement of economical and robust processes to the manufacture of new APIs at industrial scale, mainly related to remarkably potent APIs (HPAPIs) from anticancer to respiratory medicines.
Two extraordinary FDA approvals have heralded a manufacturing paradigm shi' in the direction of continuous manufacturing. The 1st was for Vertex’s Orkambi (lumaca'or/ivaca'or for cystic fibrosis) in 2015 as the initial New Drug Software (NDA) acceptance for utilizing a ongoing manufacturing engineering for manufacturing. A 4,000-square-foot ongoing manufacturing facility was in-built Boston for this goal. The second FDA approval was for Johnson & Johnson’s Prezista (darunavir for HIV) in 2016 as the primary NDA supplement acceptance for switching from batch manufacturing to steady manufacturing.
Some feedback from compact firms were being vital of your requirement that independent personnel complete structure testimonials and mentioned they must employ outdoors engineers for this endeavor. In the ultimate rule FDA will allow higher versatility and states the unbiased personnel might be individual(s) who do not have direct duty for the design stage becoming reviewed. Hence, team staff (together with engineers engaged on other elements in the machine and nonengineering staff) can execute design critiques.
• The active ingredients must have equal prominence Together with the brand title on the front/major panel of the label.
“One of several first issues they questioned us was ‘The amount of manufacturers of pharmaceuticals are there?’ ” DiLoreto recollects. The BPTF has worked While using the FDA to establish a database of drug manufacturing facilities and to aid the agency in much better comprehending how the availability chain will work.
Celebrity Then and Now
 Michelle Pfeiffer Then & Now!
Michelle Pfeiffer Then & Now!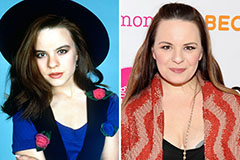 Jenna Von Oy Then & Now!
Jenna Von Oy Then & Now! Seth Green Then & Now!
Seth Green Then & Now! Loni Anderson Then & Now!
Loni Anderson Then & Now! Dolly Parton Then & Now!
Dolly Parton Then & Now!